


















 BLOGS
BLOGS  NEWSROOM
NEWSROOM  CASE STUDIES
CASE STUDIES  WEBINARS
WEBINARS  PODCASTS
PODCASTS  ASSET HUB
ASSET HUB  EVENT CALENDAR
EVENT CALENDAR 


























TL;DR
Two federal regimes now apply to US clinical decision support products: FDA Section 3060 (updated January 2026) and ONC HTI-1 (in force February 2024). The key January 2026 change: single-recommendation CDS can now qualify for the Non-Device exemption. AI/ML CDS that processes medical images or IVD signals is regulated as SaMD and does not qualify. HTI-1 adds transparency obligations for any CDS inside certified health IT, even when FDA exempts the software. Both regimes must be mapped early, regulatory pathway is a design decision.
1. Two Regulatory Regimes a US CDS Builder Now Faces
Two federal regimes now apply to a US digital health company shipping a clinical decision support product. The FDA’s Section 3060 framework, codified at 21 USC 360j(o), was sharpened by the January 6, 2026 revised guidance (final guidance formally published March 11, 2026, replacing the September 2022 version). The Office of the National Coordinator’s HTI-1 transparency rule, finalized at 89 FR 1192 (Doc 2023-28857) and effective February 8, 2024, governs Decision Support Interventions running inside ONC-certified health IT.
The two regimes overlap. A single CDS product can be FDA-exempt under Section 3060 AND subject to HTI-1 transparency requirements when it runs inside Epic, Oracle Health, or any other certified EHR. Most law-firm content addresses one regime at a time, which is fine for legal teams; product teams need the integrated picture earlier than legal teams enter the conversation, because regulatory pathway is a design decision, not a downstream review.
Market context. As of September 2025 the FDA had authorized 1,356 AI-enabled medical devices, and 1,039 of them, seventy-seven percent, are radiology products. The cleared cohort grows monthly. The exempt cohort, by definition, does not appear on any FDA list, but it is much larger and includes most rule-based medication safety, preoperative readiness, and protocol-matching CDS that surfaces recommendations without processing images or signals.
2. Is My CDS App FDA-Regulated? (Quick Answer)
Most rule-based CDS that surfaces a recommendation to a clinician, without processing medical images, IVD signals, or signal-acquisition patterns, qualifies for the Section 3060 Non-Device CDS exemption when the clinician can independently review the basis for the recommendation. AI/ML CDS that processes medical images, IVD signals, or acquisition patterns falls outside the exemption and is regulated as Software as a Medical Device under the FDA 510(k) or De Novo pathway.
The answer in three lines. The four-criteria test in the next section explains how the FDA actually evaluates each product. The single-recommendation enforcement discretion added January 2026 is the most consequential design lever buyers can apply.
3. The Section 3060 Four-Criteria Test, Sharpened in January 2026
Section 3060 of the 21st Century Cures Act, enacted December 13, 2016, amended the FD&C Act to add §520(o)(1)(E) excluding certain CDS software functions from the medical-device definition. The FDA published its first final guidance interpreting the exemption in September 2022. The January 6, 2026 revision (final guidance formally published March 11, 2026 on fda.gov) replaces it.
The four criteria, all of which must hold for the exemption to apply:
- Criterion 1. The software is not intended to acquire, process, or analyze a medical image, signal from an in vitro diagnostic device, or pattern or signal from a signal-acquisition system. The FDA interprets “process or analyze” broadly: software that “assesses or interprets the clinical implications or clinical relevance of a signal, pattern, or medical image” is treated as image/signal-processing software for this purpose. Reading pixels, ECG waveforms, or pulse-ox traces and producing a clinical interpretation crosses the line.
- Criterion 2. The software is intended for the purpose of displaying, analyzing, or printing medical information about a patient or other medical information.
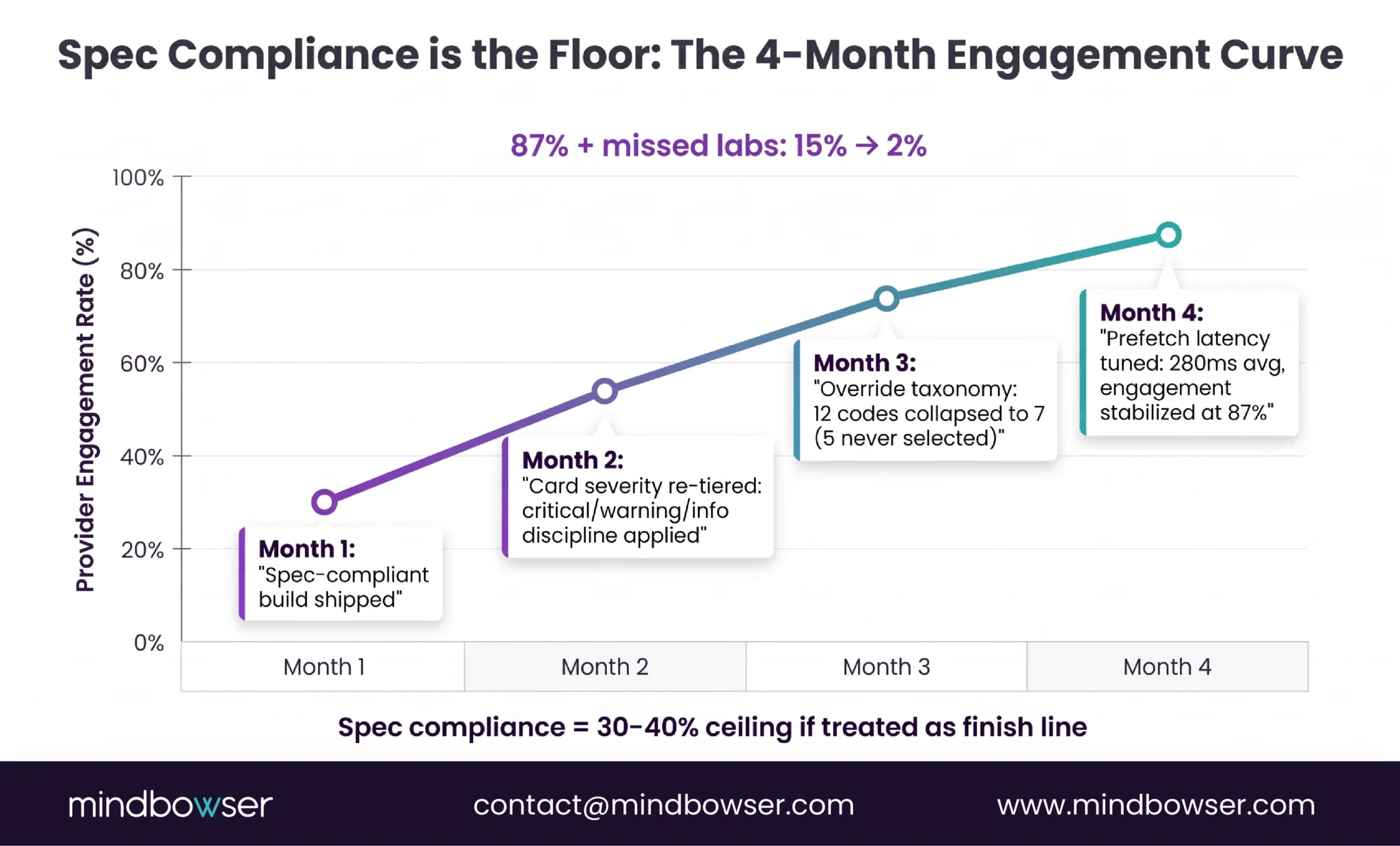
- Criterion 3. The software is intended for the purpose of supporting or providing recommendations to a healthcare professional about prevention, diagnosis, or treatment of a disease or condition. This is where the January 2026 revision changed the regulatory surface. Per DLA Piper’s January 2026 client alert, Covington & Burling’s 5 Key Takeaways, and Faegre Drinker’s analysis, FDA now intends to exercise enforcement discretion under Criterion 3 for software that provides a single recommendation. The 2022 guidance had narrowly required CDS to surface a list of options; the 2026 guidance permits a single clinically appropriate recommendation, when all other criteria are met. Hyman, Phelps & McNamara’s FDA Law Blog confirmed this is the most consequential operational change in the revision.
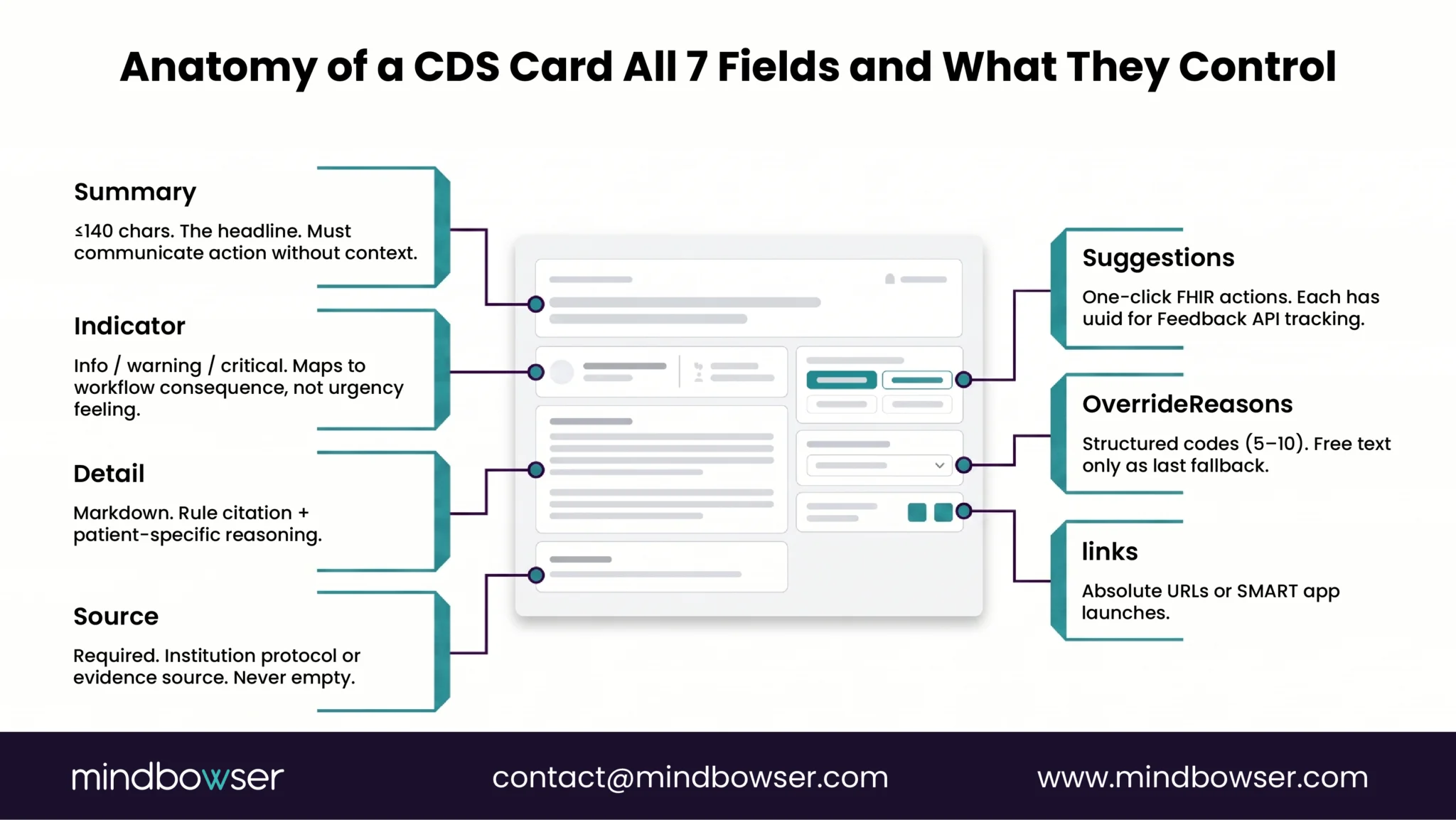
- Criterion 4. The software is intended to enable the healthcare professional to independently review the basis for the recommendation. This is the transparency criterion. The clinician must be able to see the rule, source, evidence, or model rationale that produced the output and decide whether to act on it.
The practical implication of the January 2026 change. Under the 2022 guidance, a medication-safety CDS that surfaced “recommend INR check in 48 hours given amiodarone-warfarin combination” faced regulatory ambiguity because it produced one specific recommendation rather than a list of alternatives. The 2026 revision resolves the ambiguity in favor of single-recommendation CDS as long as the underlying rule and the clinician-reviewable basis are explicit. The CITI Program’s compliance summary describes the change as the FDA “sharpening boundaries and avoiding time-critical black-box reliance.” Both directions favor product designs that expose their reasoning to the user.

4. Where CDS Crosses Into SaMD: The Image and Signal Line
Honest read of where most products cross the boundary. Criterion 1, the no-image-no-signal-no-IVD-pattern requirement, is the most common trip wire. Any CDS that processes pixels, waveforms, or signal-acquisition data and produces a clinical interpretation falls outside Section 3060 and is regulated as Software as a Medical Device.
The line in concrete examples.
| Stays exempt under Section 3060 | Crosses into SaMD |
|---|---|
| Medication safety CDS using structured drug name + allergy + dose + eGFR value | Radiology AI reading CT, MRI, X-ray, mammography, ultrasound, or PET imaging |
| Preoperative readiness CDS using lab values + diagnosis codes (PeriopMD pattern) | ECG-based arrhythmia classification or QT-prolongation detection |
| Evidence-based protocol matching using diagnosis + medication history | Retinal imaging diagnostics or dermatology AI from photos |
| Contrast-safety guidance using eGFR + weight + age | ICU vital-sign deterioration detection from continuous telemetry |
| Risk scoring from structured FHIR data (BirthModel-style, no image) | Pathology AI reading whole-slide images |
Most of the 1,039 FDA-cleared radiology AI products on the September 2025 device list cross the line by design and operate as 510(k)-cleared SaMD. Aidoc, Viz.ai, RapidAI, and the OEM-bundled AI products are all cleared devices. The integration layer that consumes their already-cleared output (covered in our radiology clinical decision support write-up) is generally exempt orchestration; the cleared models themselves are not.
The pathway when SaMD applies. The 510(k) pathway works when a predicate device exists (most cleared radiology AI uses 510(k)). De Novo applies for novel low-to-moderate-risk classifications without a predicate. PMA applies for high-risk software (rare for CDS). Frier Levitt’s developer-facing summary walks through which pathway typically maps to which CDS scope.
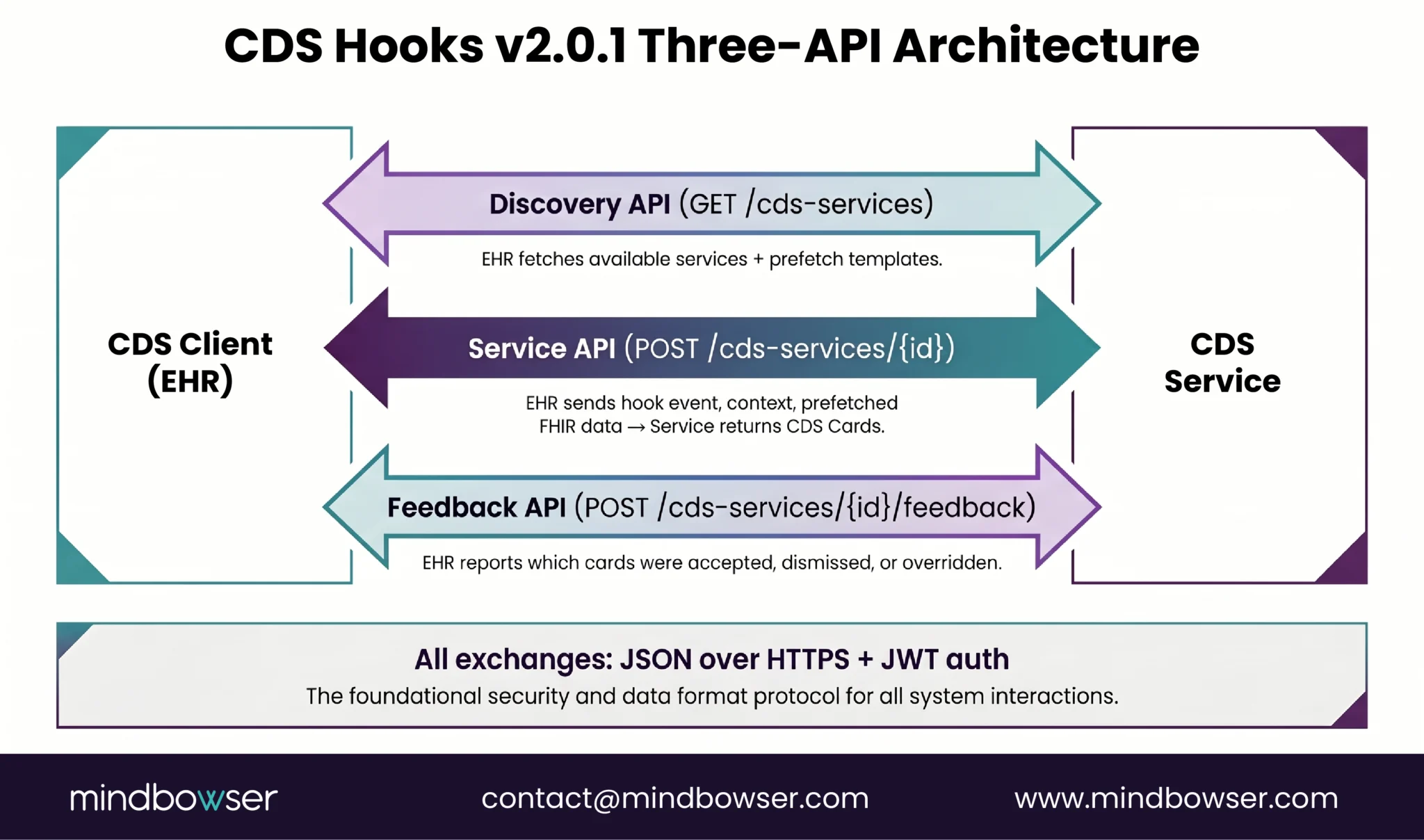
5. ONC HTI-1: The Layer That Applies Even When FDA Exempts You
The regulation Series B+ digital health builders most commonly underweight. The HTI-1 Final Rule, published January 8, 2024 at 89 FR 1192 and effective February 8, 2024, introduced first-of-its-kind transparency requirements for AI and other predictive algorithms in ONC-certified health IT.
Per Mintz’s January 2024 analysis and AHIMA’s regulatory resource, the rule defines two DSI categories.
Evidence-based DSIs. Rule-based logic derived from established clinical evidence. Examples: drug-drug interaction alerts based on standard pharmacology references, preventive-care reminders based on USPSTF guidelines, contrast-safety rules based on eGFR thresholds.
Predictive DSIs. Technology that supports decision-making based on algorithms or models that derive relationships from training data and produce a prediction, classification, recommendation, evaluation, or analysis. Examples: readmission risk scores trained on institutional data, sepsis prediction models, deterioration scores derived from machine learning over structured data.
The transparency obligations differ by category. Evidence-based DSIs require 13 source attributes (purpose, intended use, evidence base, validation, etc.). Predictive DSIs require 31 source attributes, covering developer information, intended use and population, training data details, fairness assessment methodology, validation methodology, ongoing performance metrics, and more. The disclosure framework uses the FAVES criteria (Fair, Appropriate, Valid, Effective, Safe), which certified health IT must use to evaluate Predictive DSIs.
Beyond disclosure, HTI-1 requires Intervention Risk Management practices for Predictive DSIs. Per Hooper Lundy’s takeaway summary, developers must apply Risk Analysis covering “validity, reliability, robustness, fairness, intelligibility, safety, security, and privacy,” plus Risk Mitigation practices and Governance for how data are acquired, managed, and used.
Compliance timeline. Health IT developers were required to update certified health IT to meet HTI-1 by December 31, 2024, with ongoing maintenance obligations starting January 1, 2025. As of 2026 the rule is fully in operational phase; compliance is not optional and audits are occurring.
The trap for Series B+ builders is the layered-regime question. A CDS product can be FDA-exempt under Section 3060 (rule-based, no images, clinician-reviewable basis) AND subject to HTI-1 transparency obligations (running inside Epic, Oracle Health, Athena, or any other certified EHR). The Section 3060 exemption does not exempt a product from HTI-1. The two regimes apply concurrently. A predictive ML model that derives output from structured data without processing images can sit inside Section 3060 cleanly, but as a Predictive DSI under HTI-1 it owes 31 source attributes, FAVES disclosure, and IRM documentation.

6. Designing Exempt-by-Design: PeriopMD vs BirthModel as Pattern References
Build perspective. The product design implications of the regulatory map are practical, not abstract. Two examples from our portfolio show how the design decisions land at opposite ends of the same exempt corridor.
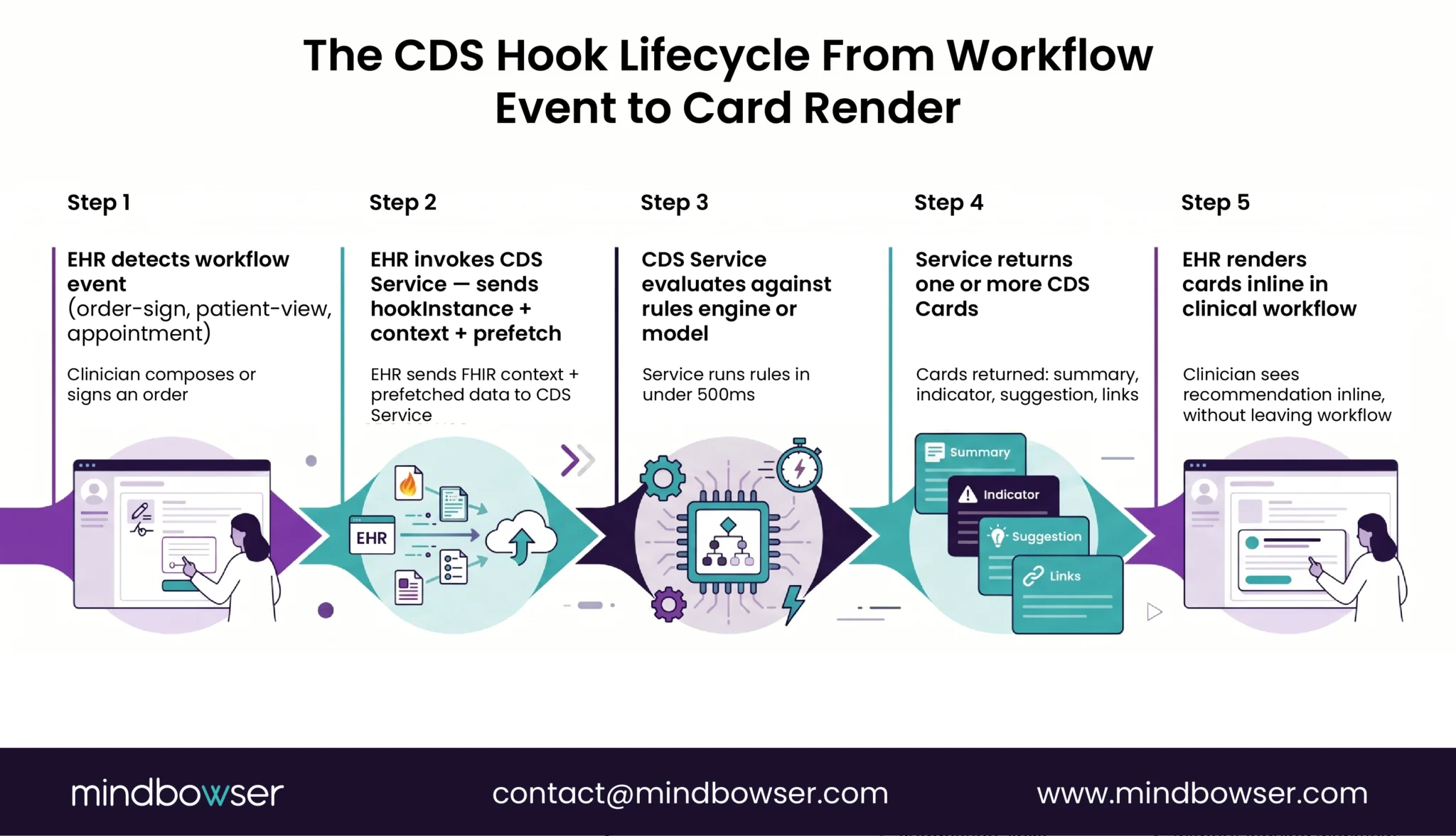
PeriopMD as exempt-by-design reference. A rule-based CDS Hooks engine. CDS Card surfaces preoperative-lab recommendations triggered by order-sign events in Epic. The rules engine evaluates against patient context (existing labs, allergies, dose, age) and returns a structured recommendation: “missing CBC for this surgical category; recommend before clearance.” The clinician (anesthesiologist) reviews the basis, sees which lab is missing and why the protocol calls for it, and decides whether to act. PeriopMD reached 87 percent provider engagement on this design.
The Section 3060 fit. Criterion 1 holds (no image or signal processing). Criterion 2 holds (displays medical info about the patient). Criterion 3 holds (recommendation to HCP about prevention/diagnosis/treatment of a condition). Criterion 4 holds (clinician reviews the basis). The January 2026 single-recommendation enforcement discretion makes the design even cleaner: the recommendation can be “this specific lab missing” rather than artificially listing alternatives to qualify under the 2022 reading.
BirthModel as boundary case. AI/ML predictive model integrated into Epic via SMART on FHIR. The model produces a probabilistic prediction (delivery timing accuracy +/- 12 minutes, 83 percent prediction accuracy on the pre-admission model) from structured patient inputs. The output is a structured prediction, not an image-derived diagnosis. The model derives output from training data without processing medical images or IVD signals.
The classification picture. BirthModel sits closer to the Section 3060 boundary than PeriopMD because its output is probabilistic and ML-derived. The exemption still holds when the model qualifies under all four criteria. Criterion 1 holds (no image processing). Criterion 4 holds (the obstetric team reviews the prediction and the clinical context). But running inside an ONC-certified EHR triggers HTI-1 Predictive DSI obligations: 31 source attributes, FAVES criteria, IRM practices. FDA exempts the software; HTI-1 still requires transparency. Design discipline must cover both regimes.
The build pattern that survives both regimes:
- Source data: structured FHIR resources only (
MedicationRequest,AllergyIntolerance,Observation,ServiceRequest,Condition). Keep image and signal processing out of the CDS itself. If an imaging or signal interpretation is required, treat it as a separate cleared SaMD component the CDS consumes. - Output: clinician-reviewable recommendation with explicit basis exposed to the user (rule citation, source evidence, model rationale).
- Architecture: CDS Hooks
order-signororder-selectfor delivery (PeriopMD pattern). If predictive AI/ML is involved, document training data lineage, fairness assessment methodology, validation results, and ongoing performance metrics from the start. These artifacts feed both HTI-1 source attributes and any future SaMD pathway if the product later expands into image processing. - Documentation: Quality System Documentation file that maps each component to its regulatory classification: exempt under Section 3060, FAVES-disclosed under HTI-1, or 510(k)-cleared SaMD if applicable. Auditors trace components through the QMS, not the marketing site.
A note on what we ship and what we do not. Mindbowser does not have a packaged “regulatory readiness” accelerator. Our HealthConnect CoPilot accelerator handles the FHIR data layer, which is regulatory-neutral plumbing. Our PHISecure / SecureSphere accelerator handles PHI de-identification (relevant for HIPAA and IRB workflows but not directly determinative for FDA classification or HTI-1 disclosure). The regulatory architecture, the design-to-stay-exempt decisions, the QMS documentation, and the HTI-1 source-attribute artifacts are custom build modeled on the PeriopMD and BirthModel patterns. There is no shortcut, and saying otherwise would oversell what the accelerators do.
The pattern transfers laterally. Our medication safety CDS write-up walks through the exempt-by-design discipline applied to drug-drug interaction and dose checking. The advanced clinical decision support write-up covers the rules-engine architecture at deeper protocol depth.
What This Means for Your Build
The FDA’s January 2026 revision made it easier to design CDS that fits the Section 3060 exemption. The single-recommendation enforcement discretion under Criterion 3 removes the prior ambiguity that pushed designers to surface artificial lists of alternatives. ONC HTI-1 remains in force concurrently and applies to predictive DSIs running inside any ONC-certified EHR, regardless of FDA classification. The product teams that get this right map design decisions to both regimes early, ship Quality System Documentation that traces each component, and treat regulatory pathway as a strategic decision alongside product architecture.
Most rule-based CDS that surfaces a recommendation with clinician-reviewable basis and structured-data inputs sits comfortably inside Section 3060. Most AI/ML CDS that processes medical images, IVD signals, or acquisition patterns is regulated SaMD. Predictive ML over structured data with clinician-reviewable basis is the boundary case where design discipline pays the most. HTI-1 obligations apply across both categories whenever the product runs inside certified health IT.
If you are a VP Regulatory or CMO at a Series B+ digital health company shipping a CDS product on top of Epic or Oracle Health, the build pattern that survives both regimes is well-documented now. PeriopMD shows the exempt-by-design pattern at production scale. BirthModel shows the predictive-DSI boundary case. Both run today.
Request an Assessment. A 30-minute scoping conversation, your product architecture plus our regulatory-design pattern, no obligation. Start a Conversation.
Conclusion
Regulatory pathway is a design decision, not a launch checklist item. The January 2026 FDA update opened a meaningful door, single-recommendation CDS can now qualify for the Non-Device exemption when clinicians can independently review the basis for each recommendation. That changes the calculus for AI-driven clinical decision support that was previously stuck on Criterion 3. The ONC HTI-1 regime runs in parallel and does not care whether FDA exempts your software, if it sits inside a certified EHR, the 31 source-attribute transparency obligation and FAVES criteria apply. Map both regimes against your data inputs, output mechanism, and deployment environment before the architecture is locked. Retrofitting regulatory compliance after a product ships costs more and takes longer than designing for the exemption from the start.
Most rule-based CDS that surfaces a recommendation to a clinician without processing medical images, IVD signals, or signal-acquisition patterns qualifies for the Section 3060 Non-Device CDS exemption when the clinician can independently review the basis for the recommendation. AI/ML CDS that processes medical images (radiology AI, dermatology AI from photos, retinal imaging diagnostics) or signals (ECG-based arrhythmia classifiers, ICU telemetry deterioration detectors) crosses into Software as a Medical Device territory and is regulated under FDA 510(k), De Novo, or PMA pathway depending on risk classification and predicate availability.
The most consequential change is enforcement discretion under Criterion 3 of the four-criteria test. The September 2022 guidance had narrowly required CDS to surface a list of options to qualify for the Section 3060 exemption. The January 6, 2026 revised guidance (formally published as final guidance March 11, 2026) permits a single clinically appropriate recommendation when all other criteria are met. The revision also emphasizes transparency and avoiding time-critical reliance on opaque recommendations. Multiple law-firm analyses (DLA Piper, Covington, Faegre Drinker, Hyman Phelps) confirmed the single-recommendation discretion as the practical operational change.
It depends on data inputs and output mechanism. Predictive ML or AI that derives output from training data over structured patient data, without processing medical images or signals, can qualify for the exemption when the clinician reviews the basis for each recommendation. Image-processing AI (radiology, pathology, dermatology, retinal) and signal-processing AI (ECG, telemetry) cross into SaMD territory by design and operate as 510(k)-cleared devices. The four-criteria test applies regardless of underlying technology, whether rule-based, ML, or generative AI.
Yes if it runs inside ONC-certified health IT (Epic, Oracle Health, Athena, MEDITECH, or any other ONC-certified EHR). HTI-1 (89 FR 1192, effective February 8, 2024) introduced transparency requirements for Decision Support Interventions inside certified EHRs. Predictive DSIs require 31 source attributes covering developer info, intended use, training data, fairness assessment, validation methodology, and ongoing performance. The FAVES criteria (Fair, Appropriate, Valid, Effective, Safe) plus Intervention Risk Management practices apply concurrently with FDA classification. Compliance maintenance obligations started January 1, 2025.
Non-Device CDS qualifies for the Section 3060 exemption from FDA medical-device regulation when all four criteria hold: no image/signal/IVD-pattern processing, displays medical information, supports HCP recommendations, lets HCP review the basis. The software is not regulated as a device, no 510(k) is required, and no PMA. SaMD is software that meets the FDA’s medical-device definition because at least one criterion fails, most commonly Criterion 1, when the software processes medical images or signals. SaMD is regulated under 510(k) for predicate devices, De Novo for novel low/moderate-risk classifications, or PMA for high-risk (rare in CDS).

