


















 BLOGS
BLOGS  NEWSROOM
NEWSROOM  CASE STUDIES
CASE STUDIES  WEBINARS
WEBINARS  PODCASTS
PODCASTS  ASSET HUB
ASSET HUB  EVENT CALENDAR
EVENT CALENDAR 


























TL;DR
- Not all RPM software is FDA-regulated. Software that displays patient data and generates alerts for clinician review generally qualifies as exempt Clinical Decision Support (CDS) under the FDA’s 4-criteria test.
- Software that independently diagnoses conditions or provides autonomous therapeutic recommendations crosses into Software as a Medical Device (SaMD) territory and requires 510(k) or De Novo clearance.
- The January 2026 FDA guidance update broadened CDS exemptions and added enforcement discretion for prediction software (Arnold & Porter, January 2026).
- RPM hardware devices (BP cuffs, pulse oximeters, CGMs) are typically Class II cleared through 510(k). The regulatory environment for RPM software is becoming more permissive, not more restrictive.
The question we get from every CTO building an RPM platform with AI-powered alerts is the same: does this need 510(k) clearance?
The answer depends on what the software does with the data after it arrives. Displaying a patient’s blood pressure trend for a care manager to review is not a medical device function. Autonomously diagnosing hypertensive crisis and triggering a medication adjustment without clinician review is. The line between those two scenarios is where FDA classification lives.
We have navigated this boundary on multiple RPM platform builds. The conversation with regulatory counsel follows a predictable pattern: define the intended use, apply the 4-criteria CDS test, document the analysis, and determine whether you are building CDS (exempt) or SaMD (regulated). For most RPM platforms, the answer is CDS. But the analysis must happen before development, not after, because intended use determines regulatory classification, and intended use is defined by your design decisions.
The January 2026 FDA guidance update made this conversation easier. The agency broadened CDS exemptions and adopted a more permissive posture toward digital health software. Understanding what changed and what stayed the same is essential for any team building RPM technology in 2026.
When Does RPM Software Become a Medical Device?

The FDA defines Software as a Medical Device (SaMD) as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device” (FDA SaMD Overview).
The key phrase is “medical purposes.” Not all software used in healthcare is a medical device. An RPM platform that collects, displays, and routes patient vital sign data to clinicians is a clinical tool. Whether it is a regulated medical device depends on what it does with that data.
The spectrum:
- Not a device (data display). Software that receives vital sign readings from devices and displays them on a dashboard for a clinician to review. No analysis, no alerts, no recommendations. Pure data presentation. The electronic equivalent of printing out the readings and handing them to a nurse. Not regulated.
- Generally exempt (CDS). Software that analyzes vital sign data, generates alerts when readings cross thresholds, calculates risk scores, and recommends actions to a clinician who reviews the data and makes the final clinical decision. This is Clinical Decision Support. If it meets the 4-criteria test (next section), it is exempt from FDA device regulation.
- Regulated (SaMD). Software that independently diagnoses a condition (arrhythmia detection from ECG waveform without clinician review), autonomously triggers therapeutic actions (closed-loop insulin pump adjusting dosing based on CGM data), or provides treatment directives that the clinician relies on primarily without independent evaluation. This is SaMD and requires 510(k), De Novo, or PMA clearance depending on the risk classification.
Most RPM platforms sit in the CDS space. They receive device data, process it through alert logic or AI-powered risk scoring, and present the output to a care manager or physician who evaluates the recommendation and decides whether to act. The clinician remains in the loop. The software supports the decision. It does not make the decision.
The 4-Criteria CDS Exemption Test
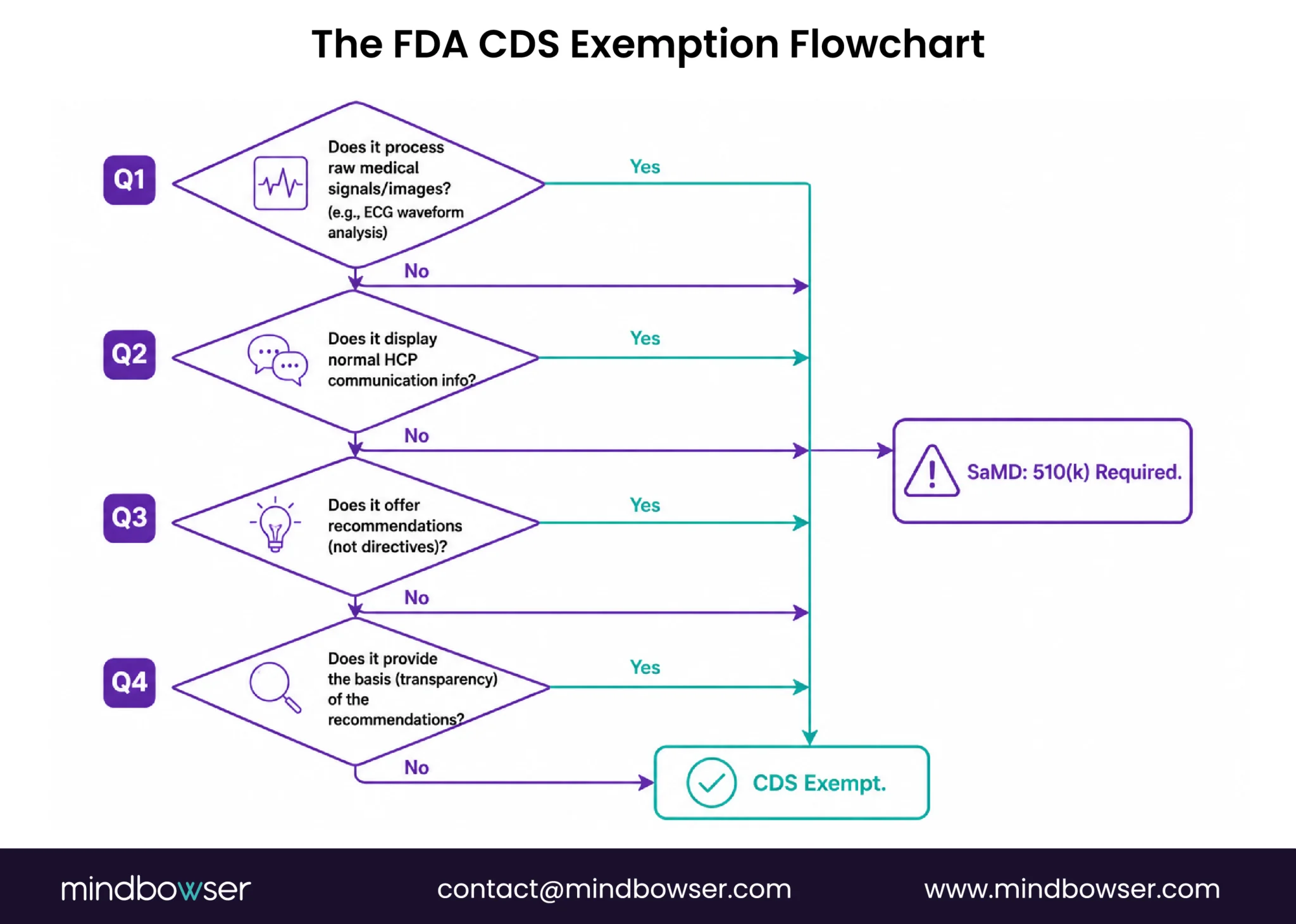
For RPM software to qualify as exempt CDS (not subject to FDA device regulation), it must meet all four criteria specified in Section 520(o)(1)(E) of the Federal Food, Drug, and Cosmetic Act (FDA CDS FAQs).
Criterion 1: Does NOT acquire, process, or analyze medical images, signals, or patterns.
This is the criterion most relevant to RPM. Displaying a blood pressure number: passes. Displaying an SpO2 value: passes. Processing an ECG waveform to detect arrhythmia: may fail this criterion because the software is analyzing a medical signal. The distinction: receiving a heart rate number from an ECG device (passes) versus analyzing the raw ECG waveform to identify rhythm patterns (may not pass).
For RPM platforms that receive processed values from devices (the device handles signal processing, the platform receives the result), this criterion is generally met. For platforms that perform their own signal analysis on raw waveform data, consult regulatory counsel.
Criterion 2: Displays, analyzes, or prints medical information normally communicated between healthcare professionals.
RPM platforms displaying vital sign trends, risk scores, and alert summaries for care team review meet this criterion. The information being communicated (vital signs, trends, alerts) is exactly the type of information HCPs normally exchange about patients.
Criterion 3: Provides recommendations (information/options) to an HCP rather than a specific output or directive.
An RPM alert that says “Patient’s SpO2 has dropped 4% from baseline; composite risk score is high; consider respiratory assessment” provides a recommendation. The clinician evaluates and decides. An RPM system that says “Administer 40mg furosemide IV now” provides a directive. The first passes criterion 3. The second does not.
Criterion 4: Provides the basis of the recommendations so the HCP does NOT rely primarily on the software to make the decision.
This is the criterion that determines whether your AI-powered alert triage is exempt or regulated. The test: does the clinician have enough information to independently evaluate the recommendation?
An alert that shows: “SpO2 dropped from patient baseline of 93.4% to 89.2% over 48 hours (deviation: -4.2%, 3.8 SD). Respiratory rate increased from baseline 16.2 to 20.1. Activity decreased 22% from 7-day average. Composite risk: high. Recommend care manager call.” That alert provides the basis. The clinician can look at the underlying data and agree or disagree with the recommendation.
An alert that shows only: “Patient risk level: HIGH. Recommended action: call patient.” That alert does not provide the basis. The clinician has no way to independently evaluate why the risk is high. They are relying primarily on the software’s assessment.
January 2026 update. The FDA now exercises enforcement discretion even when only one recommendation option is provided, as long as only one recommendation is clinically appropriate (Latham & Watkins, January 2026). Previously, providing a single recommendation (rather than multiple options) risked failing criterion 3. The updated guidance relaxes this interpretation.
What Changed in the January 2026 FDA Guidance?
On January 6, 2026, the FDA issued updated guidance documents on both Clinical Decision Support software and general wellness products. Multiple law firms published detailed analyses within days. The consensus: the regulatory environment for digital health software moved in a more permissive direction.
CDS guidance changes:
- Broader enforcement discretion for prediction software. AI/ML models that predict clinical deterioration (the exact function RPMCheck AI and Biofourmis Biovitals perform) fall more clearly under CDS exemption when the prediction is presented to a clinician with supporting data
- Single-recommendation permissibility. Previously, some interpretations of criterion 3 required presenting multiple options. The updated guidance allows a single recommendation when only one recommendation is clinically appropriate
- FDA held a Town Hall on March 11, 2026 to discuss the CDS final guidance, indicating the agency views this as an evolving policy area with industry input
General wellness guidance changes:
- Broader scope for non-invasive wearables as general wellness products exempt from regulation. This affects consumer wearables used in RPM (Apple Watch, Fitbit, Oura) when used for general wellness purposes rather than medical diagnosis
- Wearables that track activity, sleep, heart rate for wellness (not diagnosis) are more clearly exempt under the updated guidance
What did NOT change:
- SaMD that independently diagnoses conditions still requires clearance
- Devices that acquire and process medical signals (ECG waveform analysis, continuous SpO2 pattern analysis for diagnosis) still fall under FDA regulation
- The 4-criteria CDS test structure remains the same. The interpretation broadened, but the criteria did not change
- Cybersecurity requirements for connected devices remain in force
The Arnold & Porter advisory summarized it: “FDA cuts red tape on clinical decision support software and wearable products for general wellness.” For RPM builders, this means more confidence that CDS-positioned platforms are and will remain exempt.
Need Help Building FDA-ready RPM Software? Request an Assessment with Mindbowser’s Healthcare Technology Team
How Are RPM Hardware Devices Classified?
RPM hardware devices are separate from RPM software in FDA classification. The devices themselves have their own clearance status.
| Device Category | Typical FDA Class | Regulatory Pathway | Examples |
|---|---|---|---|
| Blood pressure monitors | Class II | 510(k) | Omron, Tenovi, Withings, BodyTrace |
| Pulse oximeters | Class II | 510(k) | Masimo MightySat, Nonin, Wellue |
| Continuous glucose monitors | Class II | 510(k) or De Novo | Dexcom G7, Abbott Libre 3, Stelo (De Novo for OTC) |
| ECG monitors/patches | Class II | 510(k) | Zio (iRhythm), AliveCor, Apple Watch ECG |
| Connected weight scales | Class I (exempt) or Class II | Exempt or 510(k) | Withings Body+, BodyTrace |
| Spirometers | Class II | 510(k) | Vitalograph, NuvoAir |
| Respiratory biosensors | Class II | 510(k) | Strados RESP Biosensor |
| Smart inhalers | Class II | 510(k) | Propeller Health, Teva DigiHaler |
Important terminology distinction: “FDA-cleared” means the device went through the 510(k) pathway (demonstrated substantial equivalence to a predicate device). “FDA-approved” means the device went through the Premarket Approval (PMA) pathway, which is reserved for Class III high-risk devices. Most RPM devices are cleared, not approved. The distinction matters in marketing materials and compliance documentation. Claiming a device is “FDA-approved” when it is actually “FDA-cleared” is a regulatory misstatement.
For specific device models, vendor details, and company intelligence, see our remote patient monitoring devices list.
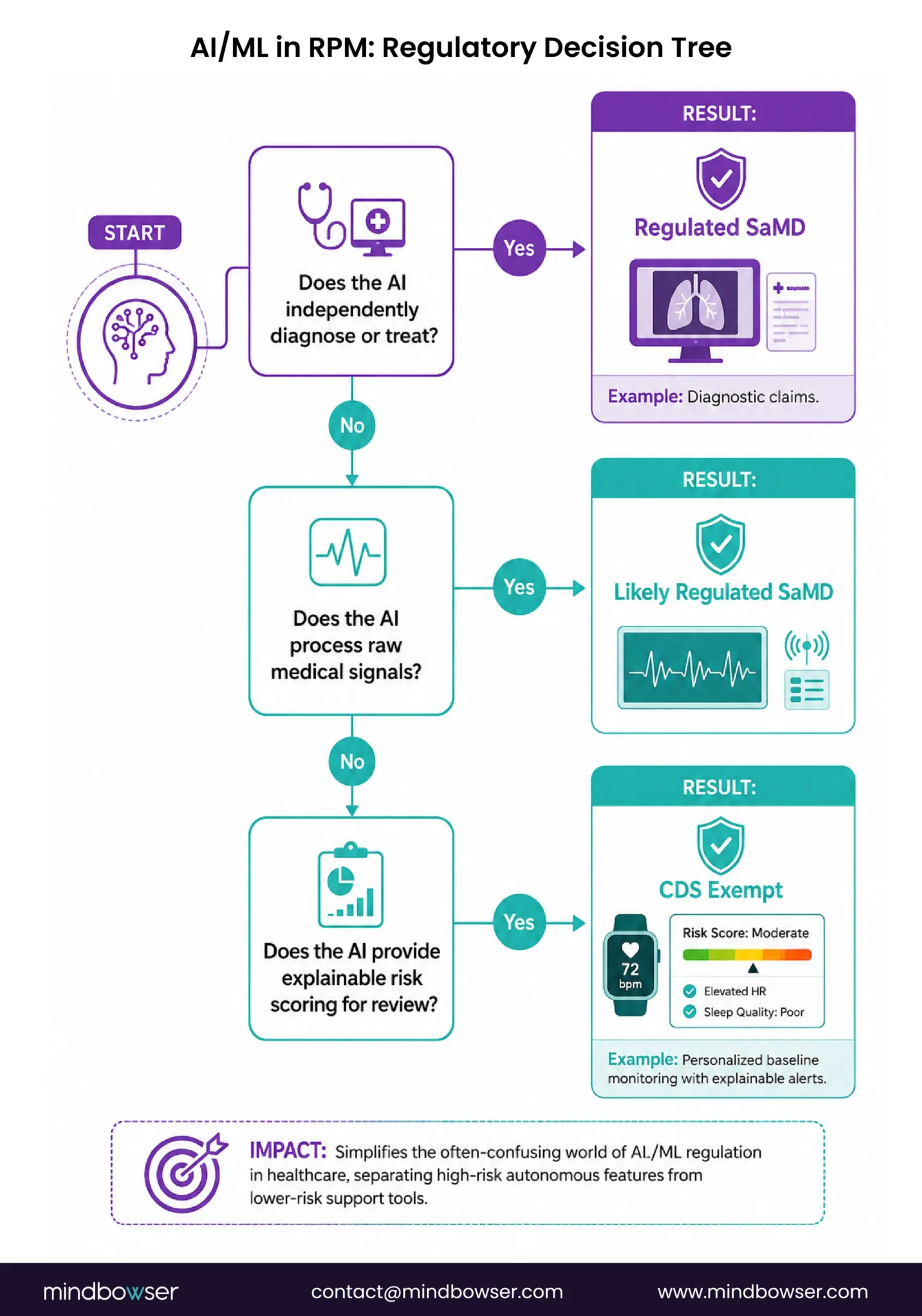
What About AI/ML in RPM Software?
The FDA maintains a public list of AI/ML-enabled medical devices that have received clearance, currently exceeding 950 devices and growing quarterly. The RPM-relevant subset includes devices like Biofourmis Biovitals (cleared for ambulatory physiologic monitoring with AI-powered deterioration prediction: 93% accuracy, 21-hour advance warning).
When RPM AI needs FDA clearance:
- Autonomous arrhythmia detection from ECG waveform analysis without clinician review
- Closed-loop therapeutic systems (CGM-to-insulin-pump without human override)
- Diagnostic claims: “this software diagnoses heart failure decompensation” (diagnostic intended use triggers SaMD classification)
- Processing raw medical signals (ECG waveforms, raw SpO2 photoplethysmography data) to produce a diagnosis
When RPM AI is exempt (CDS):
- Risk scoring with explainable reasoning presented to a clinician (the clinician reviews the data and makes the decision)
- Alert generation based on threshold or baseline deviation (the alert recommends, the clinician acts)
- Trend analysis and pattern identification surfaced for clinical review
- Predictive models that forecast risk and present the prediction with supporting data for clinician evaluation
The predetermined change control plan (PCCP) is the FDA’s framework for AI/ML devices that need to update algorithms over time. Under PCCP, a manufacturer can define a boundary for algorithm modifications in the initial clearance submission. Updates within that boundary do not require a new 510(k). This is relevant for RPM AI that learns and improves over time (personalized baselines, for example).
RPMCheck AI regulatory positioning. RPMCheck AI operates as CDS: it generates explainable alerts with underlying data visible to the clinician, who makes the final clinical decision. It is not FDA-cleared. Under current CDS guidance (including the January 2026 update), clearance is not required for this use case. Biofourmis pursued 510(k) clearance for Biovitals as a competitive differentiator and to support specific clinical claims. The clearance was a strategic choice, not a regulatory requirement for the CDS function.
For the full ML architecture behind personalized baselines and how regulatory classification applies, see our personalized baselines guide.
What Are the Cybersecurity Requirements?
The FDA’s 2025 premarket cybersecurity guidance applies to all connected medical devices, including RPM devices that transmit patient data over networks. This is device-level security (the FDA’s domain), separate from data-level security (HIPAA’s domain, covered in our HIPAA compliance checklist).
Requirements for RPM device manufacturers:
- Software Bill of Materials (SBOM). Manufacturers must provide a list of all software components in the device, including open-source libraries, third-party modules, and version numbers. This allows downstream users (health systems, RPM platform companies) to assess vulnerability exposure when a new CVE is published.
- Threat modeling and risk assessment. Cybersecurity risks must be identified and addressed during the design phase, not as an afterthought. The FDA expects “secure by design” development where threat modeling, penetration testing, and security architecture are part of the engineering process.
- Vulnerability disclosure policy. Manufacturers must have a documented process for receiving, evaluating, and publicly disclosing security vulnerabilities. If a researcher finds a flaw in a Tenovi BP cuff’s firmware, there must be a clear channel to report it and a documented timeline for remediation.
- Security patch mechanism. Connected devices must have the ability to receive firmware updates for security patches. A device deployed in a patient’s home that cannot be updated when a vulnerability is discovered is a device that becomes a permanent security risk.
- End-of-life plan. Manufacturers must define what happens when they stop supporting a device (no more security patches, no more firmware updates). RPM programs deploying devices should track device model versions and replace end-of-life devices proactively.
- What this means for RPM platform builders: these cybersecurity requirements are the device manufacturer’s responsibility, not yours. But you should verify your device vendors meet them. Ask for the SBOM. Ask about the vulnerability disclosure policy. Ask about the patch mechanism. If a vendor cannot provide these, reconsider the vendor relationship. A device with a known unpatched vulnerability in a patient’s home is both an FDA compliance issue (for the manufacturer) and a HIPAA exposure (for you).
PHISecure addresses the HIPAA data handling layer (encryption at rest and in transit, access controls, audit logging). FDA cybersecurity addresses the device security layer. Both layers apply simultaneously to a connected RPM system. One does not substitute for the other.
A Practical Compliance Checklist for RPM Builders

For RPM Platform Software
- Determine CDS exemption status. Apply the 4-criteria test to your intended use. If all four criteria are met, document the analysis and keep it on file. No FDA submission is needed, but the documentation protects you in an audit
- If software processes raw medical signals (ECG waveform analysis, SpO2 photoplethysmography pattern analysis): consult regulatory counsel. Criterion 1 may not be met, potentially classifying your software as SaMD
- If software provides autonomous therapeutic recommendations (medication dosing, treatment directives without clinician review): this is SaMD. Begin 510(k) pathway analysis
- Document your intended use statement clearly. CDS intended use (“provides clinical decision support for care team review”) differs from diagnostic intended use (“diagnoses cardiac arrhythmia”). The intended use statement is the primary driver of regulatory classification
- If using AI/ML: evaluate whether the predetermined change control plan (PCCP) framework applies. If your AI learns and updates over time, PCCP may be relevant for future regulatory needs even if current function is CDS-exempt
- Review the January 2026 CDS guidance update. Confirm your interpretation aligns with the broadened exemptions, particularly for prediction software and single-recommendation scenarios
For RPM Device Procurement
- Verify each device’s FDA clearance status. Obtain the 510(k) clearance number, classification code, and cleared indications for use. Do not rely on vendor claims of “FDA-approved” without verification
- Request the SBOM from each device manufacturer (required under 2025 cybersecurity guidance)
- Verify vulnerability disclosure policy. The manufacturer should have a documented process for reporting and remediating security vulnerabilities
- Confirm security patch mechanism. How does the device receive firmware updates? Is it automatic or manual? What is the typical patch timeline?
- Track device model versions. Maintain a registry of which device model and firmware version each patient is using. When a manufacturer issues an end-of-life notice, plan replacement proactively
- Maintain clearance documentation. Keep copies of 510(k) clearance letters, classification codes, and indications for use for each device type in your program. This documentation is required for compliance audits
The Line Moved in Your Favor
The January 2026 FDA guidance broadened CDS exemptions, added enforcement discretion for prediction software, and expanded the general wellness category for consumer wearables. The regulatory environment for RPM software is becoming more permissive.
Most RPM platforms, those that display patient data, generate alerts with explainable reasoning, calculate risk scores, and present recommendations to clinicians who make the final decision, qualify as exempt CDS under the 4-criteria test. This includes platforms with AI-powered alert triage, personalized baseline monitoring, and composite risk scoring, as long as the clinician reviews the underlying data and makes the clinical judgment.
The line to watch is when AI in RPM moves from supporting clinician decisions to making clinical decisions autonomously. An AI that says “this patient is deteriorating, here is the evidence, consider calling them” is CDS. An AI that says “this patient needs furosemide 40mg IV, administering now” is SaMD. The difference is clinician review and control.
Early in our RPM work, we were overly cautious about FDA classification, assuming any software that generated clinical alerts needed clearance. After working through the CDS criteria with regulatory counsel on three builds, we learned that the exemption is broader than most CTOs realize. The January 2026 update made it broader still. The analysis should happen early in the design phase (intended use determines classification), but for most RPM platforms, the outcome is CDS-exempt.
If you are building RPM software and unsure whether it needs 510(k) or qualifies as CDS, the analysis usually takes one meeting with your regulatory counsel and our technical team. The classification is determined by intended use and software function, both of which are defined by your design decisions. We can help frame the technical architecture in a way that supports the CDS classification when the clinical function permits it.
Not necessarily. RPM software that qualifies as Clinical Decision Support (CDS) under the FDA’s 4-criteria test is generally exempt from device regulation. The software must: (1) not process medical images/signals/patterns, (2) display information communicated between HCPs, (3) provide recommendations rather than directives, and (4) show the basis so clinicians don’t rely primarily on the software. Most RPM platforms that generate alerts for clinician review meet all four criteria. Software that independently diagnoses or provides autonomous treatment recommendations is classified as SaMD and requires 510(k) or De Novo clearance.
FDA-cleared means the device went through the 510(k) pathway, demonstrating substantial equivalence to an existing device. This applies to most RPM devices (Class II): BP monitors, pulse oximeters, CGMs, ECG monitors. FDA-approved means the device went through Premarket Approval (PMA), a more rigorous pathway for Class III high-risk devices. Almost all RPM devices are cleared, not approved. Using “FDA-approved” for a 510(k)-cleared device is a regulatory misstatement that should be avoided in marketing and compliance documentation.
The FDA’s test for determining whether software qualifies as exempt Clinical Decision Support rather than a regulated medical device. All four criteria must be met: (1) software does not acquire, process, or analyze medical images, signals, or patterns, (2) software displays/analyzes medical information normally communicated between HCPs, (3) software provides recommendations to an HCP rather than specific directives, (4) software provides the basis of recommendations so the HCP does not rely primarily on the software. If all four are met, the software is exempt. If any criterion fails, the software may be classified as SaMD.
It depends on the function. AI that generates risk scores with explainable reasoning (showing which vitals deviated, by how much, from what baseline) for clinician review is generally CDS-exempt. AI that independently diagnoses conditions (arrhythmia detection from raw ECG without clinician review) or triggers autonomous therapeutic actions is SaMD requiring clearance. Biofourmis Biovitals pursued 510(k) clearance as a competitive differentiator. RPMCheck AI operates as CDS (explainable alerts, clinician makes final decision) and does not require clearance under current guidance. The January 2026 FDA update broadened enforcement discretion for prediction software, making CDS positioning more clearly permissible.
The FDA’s 2025 premarket cybersecurity guidance requires connected medical devices to: provide a Software Bill of Materials (SBOM), include threat modeling and risk assessment during development, maintain a vulnerability disclosure policy, support security patch mechanisms for post-market updates, and define end-of-life plans. These requirements apply to device manufacturers, not RPM platform software. However, RPM platform builders should verify their device vendors meet these requirements by requesting SBOMs, vulnerability disclosure policies, and patch mechanisms.
Three changes: (1) Broader CDS enforcement discretion for prediction software, meaning AI that predicts clinical deterioration and presents predictions to clinicians is more clearly exempt from device regulation. (2) Single-recommendation permissibility, allowing CDS to provide one recommendation when only one is clinically appropriate (previously, some interpretations required multiple options). (3) Expanded general wellness scope for non-invasive consumer wearables, meaning wearables used for wellness tracking (not diagnosis) are more clearly exempt. The 4-criteria CDS test structure did not change. The interpretation became more permissive.


